Bilim insanları Rett sendromunda sızdıran beyin kan damarlarının genetiğini keşfetti
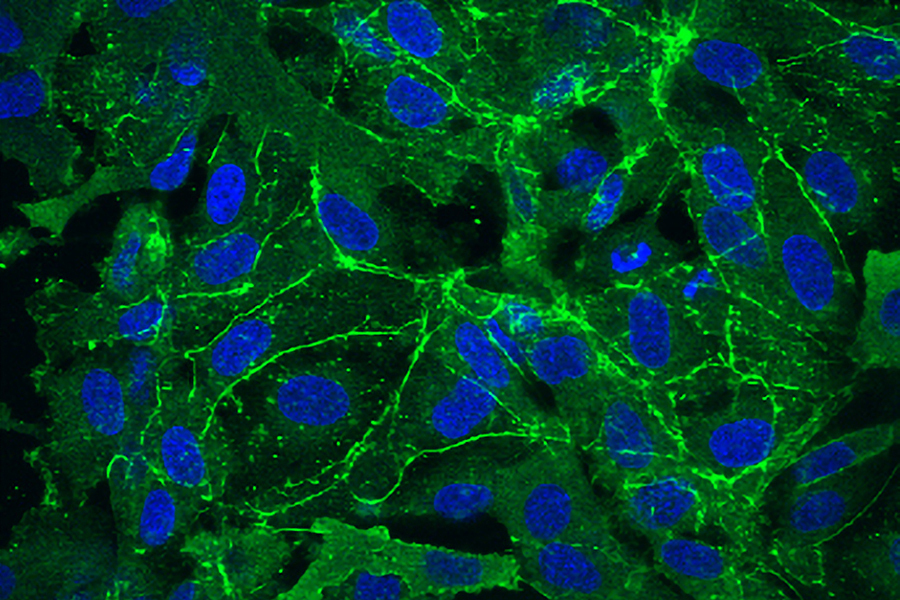
MIT araştırmacılarının çalışması, sorunun aşırı mikroRNA ifadesine yol açan genetik mutasyonlardan kaynaklandığını göstererek potansiyel bir tedaviye işaret ediyor.
MIT araştırmacılarının çalışması, sorunun aşırı mikroRNA ifadesine yol açan genetik mutasyonlardan kaynaklandığını göstererek potansiyel bir tedaviye işaret ediyor.
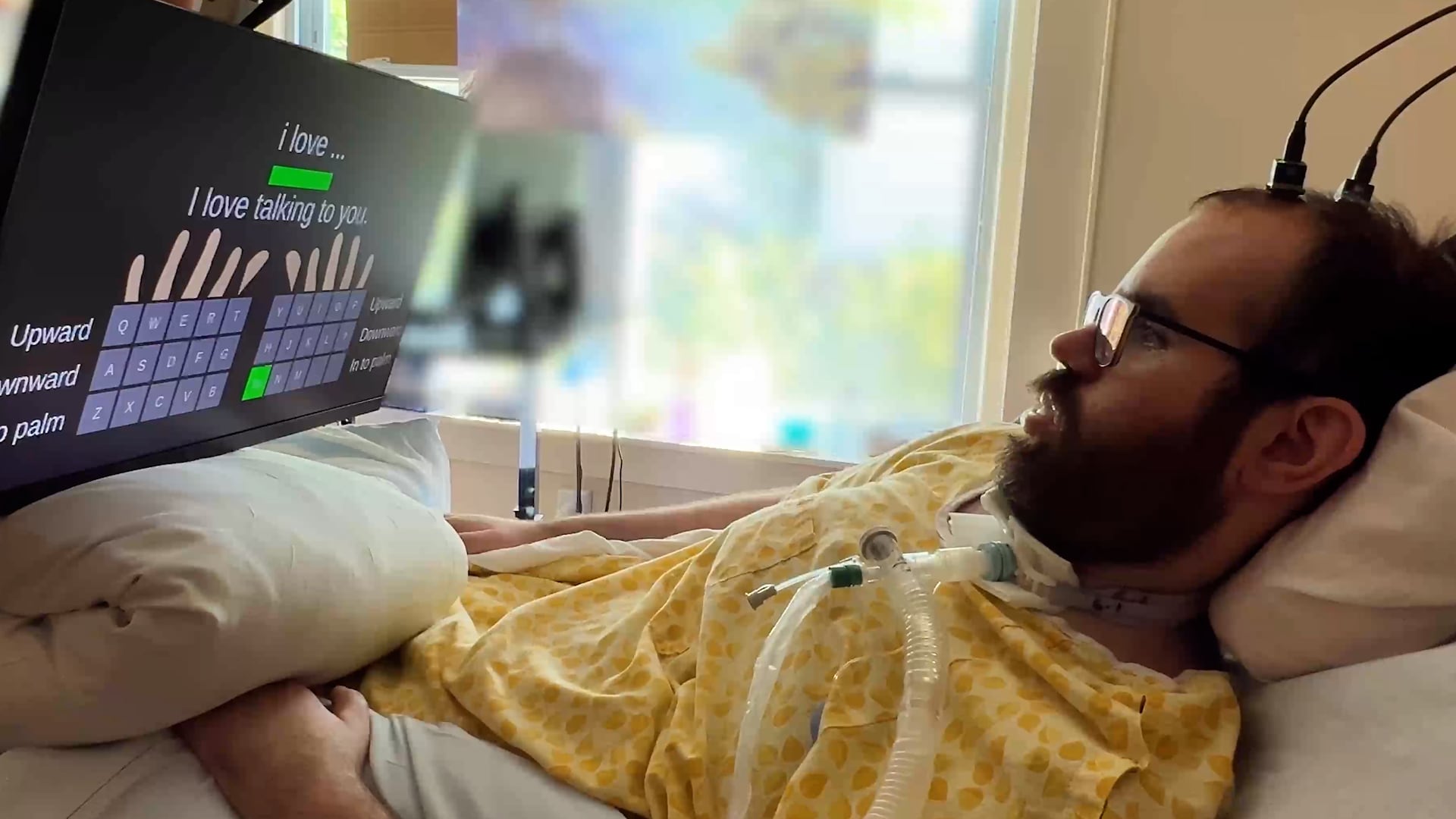
Tetraplejik bir hasta, ortalama hızlara benzer hızlar elde etti ve neredeyse hatasızdı.
 Bilim & Teknoloji
Bilim & Teknoloji Hata, giriş yapmış kullanıcıların diğer şirketlerin bilgilerini izinsiz olarak görüntüleyip düzenleyebilmesine neden oldu.
 Bilim & Teknoloji
Bilim & Teknoloji Isı kalkanları, bir uzay aracının atmosfere girdiğinde yüzeyini ve yükünü korumak için tasarlanmıştır. Illinois Üniversitesi Urbana-Champaign'deki Grainger Mühendislik Koleji'ndeki havacılık mühendisleri, Dünya gibi oksijen içeren atmosferlerle, Venüs ve Satürn'ün uydularından Titan gibi azot açısından zengin atmosferler arasındaki ısı kalkanlarının işleyişinde şiddetli yıkıcı bir fark gözlemlediler.

Antik mumyaların kendine özgü kokusu, bilim insanlarının Mısır mumyalama sırlarını çözmelerine yardımcı oluyor. Mumyaların etrafındaki havada bulunan kimyasal izleri analiz eden araştırmacılar, embalsama sırasında kullanılan yağlar, reçineler, balmumu ve bitüm ile bağlantılı onlarca bileşiği belirledi. Kimyasal ipuçları, mumyalamanın binlerce yıl boyunca giderek daha karmaşık hale geldiğini ortaya koyuyor.

Bilim ve Teknoloji Haftası'nda, Milli Teknoloji Hamlesi vizyonuyla düzenlenen etkinliklere Türkiye genelinde 100 bini aşkın kişi katıldı.

Bilim insanları, güneş ışığını faydalı kimyasal enerjiye dönüştürebilen yeni nesil malzemelerin araştırmasını hızlandırabilecek güçlü bir hesaplama yöntemi geliştirdi. Çalışma, görünür ışığı emen ve hidrojen üretimi, karbondioksit dönüşümü ve hidrojen peroksit sentezi gibi reaksiyonları yönlendirebilen umut verici bir karbon nitrit malzeme sınıfı olan poliheptazin imidlerine odaklanıyor. Araştırmacılar, 53 farklı metal iyonunun bu malzemelerin yapısı ve elektronik davranışını nasıl etkilediğini analiz ederek, hangi kombinasyonların en iyi performansı göstereceğini tahmin eden bir çerçeve oluşturdu.
 Bilim & Teknoloji
Bilim & Teknoloji Bridgnorth yakınlarında çiftçilik yapan Andrew Williamson, İran savaşının fiyatları nasıl etkilediğinden endişe duyduğunu söylüyor.

Bu panel, medya kuruluşlarının canlı yayınlarını ve hava trafiği ile askeri uyarılar gibi açık veri sinyallerini entegre etmektedir.